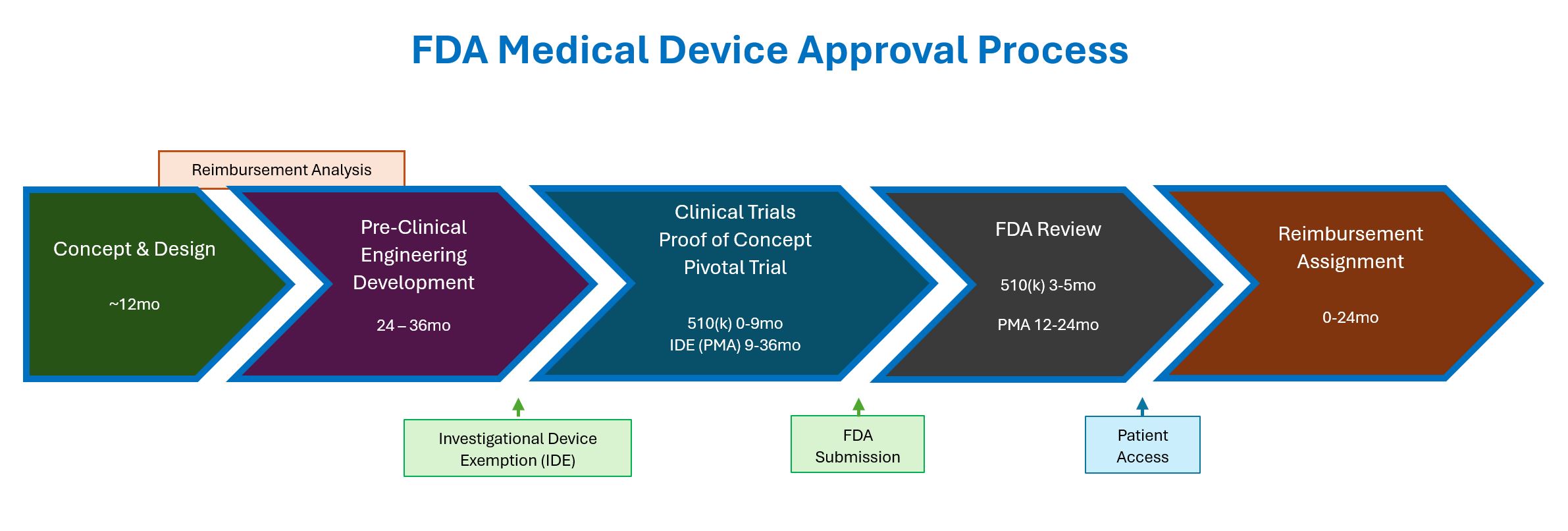
Medical Device Development
Medical Device Classification
Class I (low to moderate risk): general controls
Class II (moderate to high risk): general controls and special controls
Class III (high risk): general controls and Premarket Approval (PMA)
General Controls
- 501: Adulterated devices
- 502: Misbranded devices
- 510: Registration of producers of devices
- Establishment registration and device listing
- Premarket Notification (510k)
- Reprocessed single-use devices
- 516: Banned devices
- 518: Notifications and other remedies
- Notification
- Repair
- Replacement
- Refund
- Reimbursement
- Mandatory recall
- 519: Records and reports on devices
- Adverse event report
- Device tracking
- Unique device identification system
- Reports of removals and corrections
- 520: General provisions respecting control of devices intended for human use
- Custom device
- Restricted device
- Good manufacturing practice requirements
- Exemptions for devices for investigational use
- Transitional provisions for devices considered as new drugs
- Humanitarian device exemption
Special Controls
- Performance standards
- Postmarket surveillance
- Patient registries
- Special labeling requirements
- Premarket data requirements
- Guidelines
