
Pharmaceutical Development
TYPES OF DRUG APPLICATIONS
- Investigational New Drug (IND)
Current Federal law requires that a drug be the subject of an approved marketing application before it is transported or distributed across state lines. Because a sponsor will probably want to ship the investigational drug to clinical investigators in many states, it must seek an exemption from that legal requirement. The IND is the means through which the sponsor technically obtains this exemption from the FDA. - New Drug Application (NDA)
When the sponsor of a new drug believes that enough evidence on the drug’s safety and effectiveness has been obtained to meet FDA’s requirements for marketing approval, the sponsor submits to FDA a new drug application (NDA). The application must contain data from specific technical viewpoints for review, including chemistry, pharmacology, medical, biopharmaceutics, and statistics. If the NDA is approved, the product may be marketed in the United States. For internal tracking purposes, all NDA’s are assigned an NDA number. - Abbreviated New Drug Application (ANDA)
An Abbreviated New Drug Application (ANDA) contains data that, when submitted to FDA’s Center for Drug Evaluation and Research, Office of Generic Drugs, provides for the review and ultimate approval of a generic drug product. Generic drug applications are called “abbreviated” because they are generally not required to include preclinical (animal) and clinical (human) data to establish safety and effectiveness. Instead, a generic applicant must scientifically demonstrate that its product is bioequivalent (i.e., performs in the same manner as the innovator drug). Once approved, an applicant may manufacture and market the generic drug product to provide a safe, effective, low cost alternative to the American public. - Biologic License Application (BLA)
Biological products are approved for marketing under the provisions of the Public Health Service (PHS) Act. The Act requires a firm who manufactures a biologic for sale in interstate commerce to hold a license for the product. A biologics license application is a submission that contains specific information on the manufacturing processes, chemistry, pharmacology, clinical pharmacology and the medical affects of the biologic product. If the information provided meets FDA requirements, the application is approved and a license is issued allowing the firm to market the product.
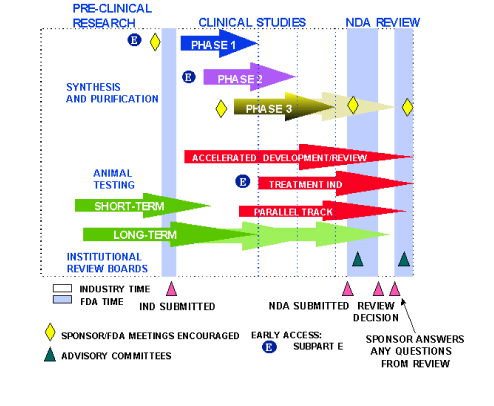
DRUG DEVELOPMENT PROCESS
- Discovery and Development – Research for a new drug begins in the laboratory.
- Preclinical Research – Drugs undergo laboratory and animal testing to answer basic questions about safety.
- Clinical Research – Drugs are tested on people to make sure they are safe and effective.
- FDA Review – FDA review teams thoroughly examine all of the submitted data related to the drug and make a decision to approve or not to approve.
- FDA Post-Market Safety Monitoring – FDA monitors all drug safety once products are available for use by the public.
DOSAGE FORMS & ROUTE OF ADMINISTRATION
Dosage forms (also called unit doses) are pharmaceutical drug products presented in a specific form for use. They contain a mixture of active ingredients and inactive components (excipients), configured in a particular way and apportioned into a specific dose.
Dosage forms vary depending on the method and route of administration, which can include many types of liquid, solid, and semisolid forms. Common dosage forms include tablets, capsules, syrups, creams, and injectables.
A combination drug (or fixed-dose combination; FDC) is a product that contains more than one active ingredient.
The route of administration (ROA) for drug delivery depends on the dosage form of the substance. Different dosage forms may be available for a particular drug, especially if certain conditions restrict the ROA. A specific dosage form may also be required due to issues such as chemical stability or pharmacokinetic properties.
CHOOSING A DOSAGE FORM
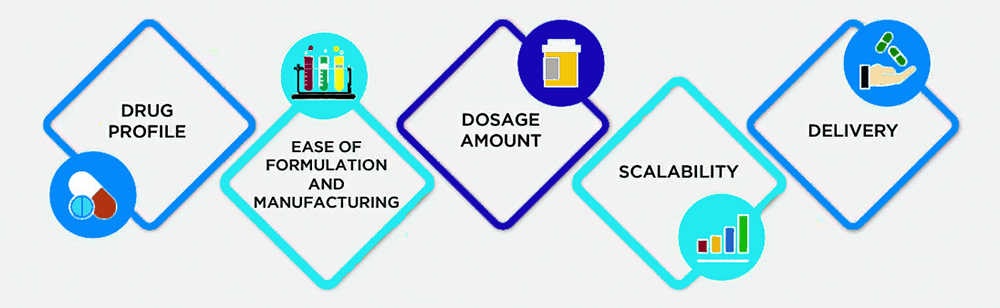
The proper design and formulation of a dosage form requires consideration of the physical, chemical, and biological characteristics of all of the drug substances (active pharmaceutical ingredients (APIs)) and pharmaceutical ingredients (excipients) to be used in manufacturing the drug product. The API and excipients utilized must be compatible and produce a drug product that is stable, efficacious, palatable, easy to administer, and well tolerated. Formulation factors include physical properties such as particle size, crystalline structure, melting point, solubility, partition coefficient, dissolution, membrane permeability, dissociation constants, and drug stability. Successful development of a dosage form includes multiple considerations involving the drug, excipients, compliance, storage, packaging, and stability, as well as patient considerations of taste, appearance, and palatability.
CHEMISTRY MANUFACTURING & CONTROLS
Chemistry Manufacturing & Controls (CMC) is an essential part of the Drug Development Process. CMC is a term used when drug developers define their investigative drug substance, establish manufacturing methods for that drug, and develop a strategy for controlling the quality and stability of the product from early phases through post-approval production.
QUALITY By DESIGN (QbD)
Quality by design (QbD) is a concept first outlined by quality expert Joseph M. Juran in publications, most notably Juran on Quality by Design, 1992. QbD is a proactive, science-based approach to product and process development that focuses on understanding the relationships between critical process parameters (CPPs) and critical quality attributes (CQAs) to ensure a product meets its intended use. The approach aligns with the eCTD format, covering the elements required for a successful regulatory submission. By using the QbD approach, development can be accelerated and resources can be used more effectively.
The quality by design model consists of the following steps:
- Establish the project design targets and goals.
- Define the market and customers that will be targeted.
- Discover the market, customers, and societal needs.
- Develop the features of the new design that will meet the needs.
- Develop or redevelop the processes to produce the features.
- Develop process controls to be able to transfer the new designs to operations.
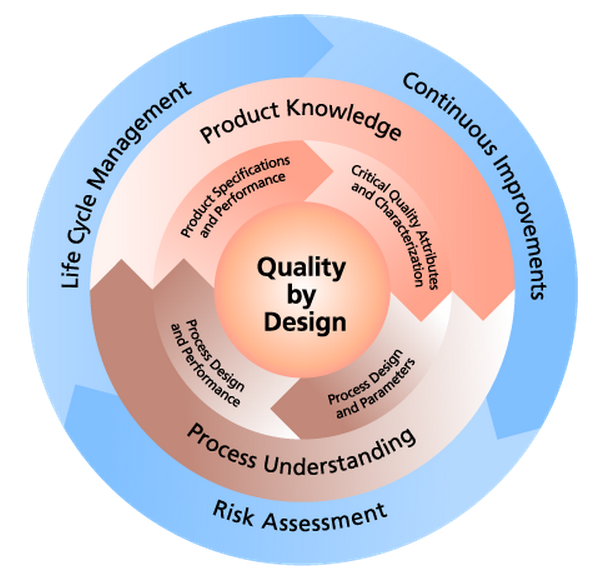
Elements of Pharmaceutical Development
- Defining the quality target product profile (QTPP) as it relates to quality, safety and efficacy, considering e.g., the route of administration, dosage form, bioavailability, strength, and stability;
- Identifying potential critical quality attributes (CQAs) of the drug product, so that those product characteristics having an impact on product quality can be studied and controlled;
- Determining the critical quality attributes of the drug substance, excipients etc., and selecting the type and amount of excipients to deliver drug product of the desired quality;
- Selecting an appropriate manufacturing process ;
- Defining a control strategy
- A systematic evaluation, understanding and refining of the formulation and manufacturing process, including;
- Identifying, through e.g., prior knowledge, experimentation, and risk assessment, the material attributes and process parameters that can have an effect on product CQAs;
- Determining the functional relationships that link material attributes and process parameters to product CQAs;
- Using the enhanced product and process understanding in combination with quality risk management to establish an appropriate control strategy which can, for example, include a proposal for a design space(s) and/or real-time release testing.
Quality Target Profile (QTP)
The quality target product profile forms the basis of design for the development of the product. Considerations for the quality target product profile could include:
- Intended use in clinical setting, route of administration, dosage form, delivery systems;
- Dosage strength(s);
- Container closure system;
- Therapeutic moiety release or delivery and attributes affecting pharmacokinetic characteristics (e.g., dissolution, aerodynamic performance) appropriate to the drug product dosage form being developed;
- Drug product quality criteria (e.g., sterility, purity, stability and drug release) appropriate for the intended marketed product.
Critical Quality Attributes (CQA)
A CQA is a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality. CQAs are generally associated with the drug substance, excipients, intermediates (in-process materials) and drug product.
Risk Assessment
Risk assessment is a valuable science-based process used in quality risk management that can aid in identifying which material attributes and process parameters potentially have an effect on product CQAs. Risk assessment is typically performed early in the pharmaceutical development process and is repeated as more information becomes available and greater knowledge is obtained.
Design Space
The relationship between the process inputs (material attributes and process parameters) and the critical quality attributes can be described in the design space.
Control Strategy
A control strategy is designed to ensure that a product of required quality will be produced consistently. The elements of the control strategy should describe and justify how in-process controls and the controls of input materials (drug substance and excipients), intermediates (in-process materials), container closure system, and drug products contribute to the final product quality. These controls should be based on product, formulation and process understanding and should include, at a minimum, control of the critical process parameters and material attributes.
Product Lifecycle Management and Continual Improvement
Throughout the product lifecycle, companies have opportunities to evaluate innovative approaches to improve product quality. Process performance can be monitored to ensure that it is working as anticipated to deliver product quality attributes as predicted by the design space. This monitoring could include trend analysis of the manufacturing process as additional experience is gained during routine manufacture. For certain design spaces using mathematical models, periodic maintenance could be useful to ensure the model’s performance. The model maintenance is an example of activity that can be managed within a company‘s own internal quality system provided the design space is unchanged. Expansion, reduction or redefinition of the design space could be desired upon gaining additional process knowledge. Change of design space is subject to regional requirements.

LINKS
FDA
- 21 CFR Part 58 Good Laboratory Practice for Nonclinical Laboratory Studies
- 21 CFR Part 312 – Investigational New Drug Application
- Dosage Forms
- Dosage Form and Route of Administration
- Sterile Drug Products Produced by Aseptic Processing – cGMP
- Guidance for Industry – Process Analytical Technology (PAT) – A Framework for Innovative Pharmaceutical Development, Manufacturing and Quality Assurance
- The Drug Development Process
- The New Drug Development and Review Process
- Guide for Industry: Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment
USP
ISPE
- Good Practice Guide: Development of Investigational Therapeutic Biological Products
- Good Practice Guide: Knowledge Management in the Pharmaceutical Industry
- Good Practice Guide: Operations Management
- Good Practice Guide: Project Management for the Pharmaceutical Industry
- Baseline Guide: Active Pharmaceutical Ingredients
- Baseline Guide: Oral Solid Dosage Forms
- Baseline Guide: Risk-based Manufacture of Pharmaceutical Products
- Guide: Biopharmaceutical Process Development and Manufacturing
- APQ Guide: Management Responsibilities and Management Review (MRR)
- APQ Guide: Process Performance and Product Quality Monitoring System (PPPQMS)