
Quality Engineering
EQUIPMENT PROCUREMENT
1. Requirement Assessment & Planning
- Identify the equipment needs based on manufacturing processes, production volume, and compliance requirements.
- Define user requirements specifications (URS), considering factors like material compatibility, automation level, and regulatory adherence (e.g., FDA, EMA, GMP).
- Conduct feasibility studies and budget planning to align with financial and operational goals.
2. Vendor Selection & Evaluation
- Research potential suppliers with experience in pharmaceutical equipment manufacturing.
- Evaluate vendors based on quality certifications (ISO 9001, GMP compliance, CE marking).
- Conduct audits or factory visits to verify the supplier’s manufacturing and quality control processes.
- Request for Quotation (RFQ) and technical proposals from shortlisted vendors.
3. Technical Review & Compliance Check
- Factory Acceptance Testing (FAT)
- Compare equipment specifications against URS and regulatory guidelines.
- Assess equipment documentation, including validation support, material certificates, and calibration details.
- Ensure the equipment meets cGMP, 21 CFR Part 11 (if software-based), and cleanroom classification requirements.
4. Procurement & Contract Finalization
- Negotiate pricing, delivery timelines, payment terms, and service agreements.
- Establish agreements for after-sales support, spare parts availability, and training.
- Issue a Purchase Order (PO) and ensure all required documentation is in place.
5. Equipment Delivery & Installation
- Coordinate logistics, customs clearance (if imported), and site readiness for installation.
- Site Acceptance Testing (SAT)
- Perform incoming inspection to verify compliance with specifications.
- Supervise installation and ensure alignment with the facility layout and safety standards.
6. Qualification & Validation
- Conduct Installation Qualification (IQ) to verify correct setup and documentation.
- Perform Operational Qualification (OQ) to ensure the equipment functions as intended under test conditions.
- Carry out Performance Qualification (PQ) by running the equipment under actual production conditions.
- Validate software systems if applicable (Computer System Validation – CSV).
7. Training & Handover
- Train operators, maintenance personnel, and quality assurance teams.
- Establish Standard Operating Procedures (SOPs) and maintenance schedules.
- Conduct final acceptance testing before full-scale production.
8. Ongoing Maintenance & Compliance
- Implement preventive maintenance schedules and periodic calibrations.
- Maintain logs for audits and inspections.
- Regularly update equipment documentation and ensure compliance with evolving regulatory standards.
VALIDATION
Validation is the process of systematically ensuring that equipment, processes, methods, and systems consistently produce results that meet predetermined quality and regulatory standards. In pharmaceutical manufacturing, validation is a critical requirement to ensure compliance with Good Manufacturing Practices (GMP) and regulatory guidelines.
Types of Validation
- Design Qualification (DQ) verifies that the equipment design meets process requirements.
- Installation Qualification (IQ) verifies that an instrument or unit of equipment being qualified (as well as its sub-systems and any ancillary systems) has been installed and configured according to the manufacturer’s specifications or installation checklist.
- Operation Qualification (OQ) involves identifying and inspecting equipment features that can impact final product quality.
- Performance Qualification (PQ) user requirements are verified as being met.
- Process Performance Qualification (PPQ) is a stage in the validation process that assess the reliability of a manufacturing process.
- Cleaning Validation confirms that cleaning procedures remove residues, contaminants, and microbial load to prevent cross-contamination.
- Computer System Validation ensures that software systems used in manufacturing and quality control are compliant with 21 CFR Part 11 regulations.
- Packaging Validation ensures that packaging materials and processes protect the drug product’s integrity, stability, and compliance with labeling requirements.
Process Validation
- Understand the sources of variation
- Detect the presence and degree of variation
- Understand the impact of variation on the process and ultimately on product attributes
- Control the variation in a manner commensurate with the risk it represents to the process and product
- Stage 1 – Process Design: The commercial manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities.
- Stage 2 – Process Qualification: During this stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing.
- Stage 3 – Continued Process Verification: Ongoing assurance is gained during routine production that the process remains in a state of control.
CALIBRATION
Calibration is the process of comparing a measuring instrument’s readings to a known standard to ensure accuracy and reliability. In pharmaceutical manufacturing, calibration is crucial for maintaining Good Manufacturing Practices (GMP) and regulatory compliance, ensuring that all instruments provide precise measurements.
- Reference Standard Selection – Use a certified standard (e.g., NIST-traceable).
- Measurement Comparison – Compare instrument readings to the reference standard.
- Adjustment & Correction – If deviations exist, adjust the instrument to align with the standard.
- Documentation – Record calibration results, date, and next calibration schedule.
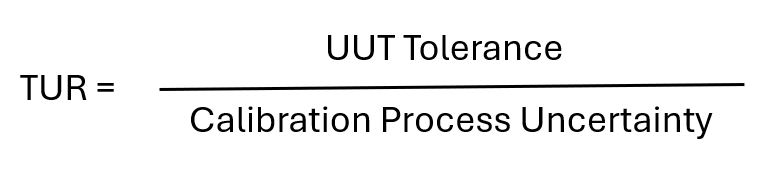
- Unit Under Test (UUT)
- Test Uncertainty Ratio (TUR)

LINKS
FDA
- 21 CFR Part 211, Subpart C – Buildings and Facilities
- 21 CFR Part 211, Subpart D – Equipment
- 21 CFR Part 211, Subpart F – Production and Process Controls, 211.105 Equipment identification
- 21 CFR Part 211, Subpart I – Laboratory Controls, 211.160(b)(4)
- 21 CFR Part 211, Subpart J – Records and Reports, 211.182 Equipment cleaning and use log.
- 21 CFR Part 820, Subpart G – Production and Process Controls, 820.70 Production and process controls
- 21 CFR Part 820, Subpart G – Production and Process Controls, 820.72 Inspection, measuring and test equipment.
- 21 CFR Part 820, Subpart G – Production and Process Controls, 820.75 Process validation
- 21 CFR Part 820, Subpart H – Acceptance Activities, 820.80(e)(5) Acceptance Records
- 21 CFR Part 820, Subpart M – Records, 820.181 Device master record
- Process Validation: General Principles and Practices
- Medical Device Development Tools (MDDT)
- Validation of Cleaning Processes (7/93)
ICH
EMA
ISO
ISPE
- GAMP 5 Guide: A Risk-based Approach to Compliant GxP Computerized Systems
- GAMP Good Practice Guide: Calibration Management
- GAMP Good Practice Guide: GxP Process Control Systems
- Good Practice Guide: Asset Management
- Good Practice Guide: Approaches to Commissioning and Qualification of Pharmaceutical Water and Steam Systems
- Good Practice Guide: Cold Chain Management
- Good Practice Guide: Containment for Potent Compounds
- Good Practice Guide: Continuous Manufacturing of Oral Solid Dosage Forms
- Good Practice Guide: Controlled Temperature Chambers
- Good Practice Guide: Critical Utilities GMP Compliance
- Good Practice Guide: Decommissioning Pharmaceutical Equipment and Facilities
- Good Practice Guide: Equipment Reliability
- Good Practice Guide: Good Engineering Practice
- Good Practice Guide: Heating, Ventilation and Air Conditioning
- Good Practice Guide: HVAC and Process Equipment Air Filters
- Good Practice Guide: Maintenance
- Good Practice Guide: Management of Engineering Standards
- Good Practice Guide: Membrane-Based WFI Systems
- Good Practice Guide: Operation Management
- Good Practice Guide: Ozone Sanitization of Pharmaceutical Water Storage and Distribution Systems
- Good Practice Guide: Packaging, Labeling and Warehouse Facilities
- Good Practice Guide: Process Gases
- Good Practice Guide: Process Validation
- Good Practice Guide: Quality Lab Facilities
- Good Practice Guide: Sampling Pharmaceutical Water, Steam and Process Gases
- Good Practice Guide: Single Use Technology
- Good Practice Guide: Standardized Methodology for the Evaluation of Pharmaceutical Airborne Particle Emissions from Containment Systems
- Good Practice Guide: Technology Transfer
- Good Practice Guide: Unique ID of Glass Primary Containers
- Baseline Guide: Active Pharmaceutical Ingredients
- Baseline Guide: Oral Solid Dosage Forms
- Baseline Guide: Sterile Product Manufacturing Facilities
- Baseline Guide: Water and Steam Systems
- Baseline Guide: Commissioning and Qualification
- Baseline Guide: Biopharmaceutical Manufacturing Facilities
- Baseline Guide: Risk-based Manufacture of Pharmaceutical Products
- Guide: Biopharmaceutical Process Development and Manufacturing
- Guide: Cleaning Validation Lifecycle – Application, Methods and Controls